Human-Derived Plasma Ancillary Materials for Cell Therapies: What Virus Inactivation Methods Should You Consider?
Human plasma is defined as “clear, straw colored, complex liquid that is 7% protein, 91% water, and 0.9% mineral salts” and can be derived from Source or Recovered Plasma. Today, human plasma is used to produce several ancillary materials such as Human AB Serum, Human Fibronectin, Human Serum Albumin (HSA), and Human Vitronectin. These materials are critical to the downstream manufacture of cell, gene, and tissue-engineered products, and are governed by USP <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products.
In the regenerative medicine and biotech industries there has been pressure from several regulatory agencies, particularly the Pharmaceuticals and Medical Devices Agency (PDMA) in Japan and Food and Drug Administration (FDA) in the US, for therapeutic companies to switch to human-derived plasma products in their manufacturing processes in order to reduce the risks of diseases and viruses such as Transmissible Spongiform Encephalopathies (TSE) and Bovine Spongiform Encephalopathy (BSE) found in bovine-derived products. The discussion has now shifted to ensure viral testing/inactivation/reduction in these human-derived plasma products.
Since ancillary materials are typically manufactured from larger plasma donor pools, there is a broad risk of viruses and bloodborne pathogens. The FDA has defined a “Five-Layer Safety Net” to address this concern, promoting donor screening, blood testing, donor referral, quarantine, and investigation – all steps that need to be considered by therapy developers sourcing these materials. The FDA, World Health Organization (WHO), International Council for Harmonization (ICH), European Medicine Agency (EMA) and others define the standard test requirements for human blood and blood components for transfusion or for further manufacturing of a drug/therapeutic product. In addition to the current safety measures in place, several therapeutic and biotech companies have requested smaller donor pools, additional virus testing, and viral inactivation/reduction to further mitigate the risk of potential virus contamination.
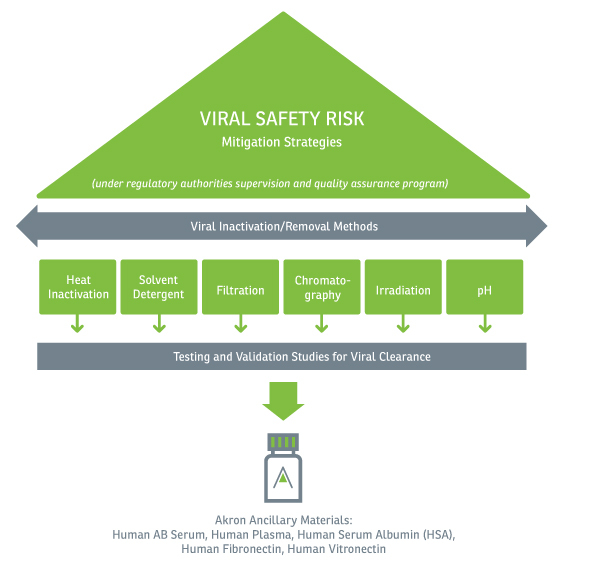
Today, there are several viral inactivation/reduction methods currently used during and after manufacture as a result of many of the virus-related infections and events during the 1980s and continuing up to the 2000s with West Nile and Zika Viruses:
- Heat inactivation
- Solvent detergent
- Filtration
- Chromatography
- Irradiation
- pH inactivation
Solvent detergent and pH inactivation are generally effective against enveloped viruses. Heat, irradiation, chromatography and filtration methods have been shown to have some effect on non-enveloped viruses, as well as enveloped viruses. In addition, more recent technologies have evolved such as membrane chromatography, filtration with orthogonal removal mechanisms or higher area filters, ultraviolet-C (UVC) light and high temperature short time (HTST) treatment.
While all methods listed above achieve some level of viral inactivation or removal, these same methods can potentially have a negative effect on the ancillary material, often stripping or inactivating critical proteins essential to the product’s performance to promote cell growth and expansion. When choosing a viral inactivation/reduction method, there is no “one-size-fits-all”. Rather, a combination of these methods should be considered and validated to reduce the risk of adventitious viruses and bloodborne pathogens in plasma derived ancillary materials and ultimately, the final drug/therapeutic product.